
Patogenesis kronis
23
PATOGENESIS KRONIS "KOR PULMONAL" PADA PPOK Paolo Palange* dan Patrizia Paoletti Departemen Kedokteran Klinis, Unit Fungsi Paru, University of Rome “La Sapienza”, Italia Abstrak: Meskipun "kor pulmonal" merupakan penyebab utama kematian pada PPOK lanjutan, tidak ada konsensus yang jelas pada definisi tersebut. Hipertensi pulmoner secara tradisional dianggap sebagai mekanisme patogenetik yang mengarah ke disfungsi jantung kanan pada PPOK, dengan edema perifer yang biasanya menjadi dihubungkan dengan kongesti vena sekunder untuk menaikkan tekanan pengisian ventrikel kanan. Kerugian pada kapiler paru dan remodeling vaskular secara sekunder terhadap penyebab hipoksia kronis, rokok asap rokok eksposisi dan inflamasi sistemik, yang terkenal disebabkan oleh disfungsi endotel dan hipertensi pulmonal pada pasien PPOK. Mekanisme lain, seperti vasokonstriksi hipoksia dan kompresi kapiler alveolar pada paru yang hiperinflasi dapat berkontribusi pada peningkatan tekanan arteri paru selama eksaserbasi PPOK, olahraga dan tidur. Dalam dekade terakhir, bagaimanapun juga, peran tekanan paru yang berlebih dalam pengembangan gagal jantung kanan telah diperdebatkan oleh kebanyakan orang, karena kurangnya korelasi yang jelas antara tekanan arteri pulmonalis dan adanya edema perifer dan / atau tanda-tanda hemodinamik gagal jantung kanan. Selain itu, adanya perubahan ginjal dan hormonal yang berbeda, menunjukkan bahwa pembentukan edema pada fase lanjutan dari PPOK, sebagian besar disebabkan oleh retensi natrium dan air yang disebabkan
-
Upload
fahmi-wahyu-rakhmanda -
Category
Documents
-
view
73 -
download
0
Transcript of Patogenesis kronis

PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
Paolo Palange dan Patrizia Paoletti
Departemen Kedokteran Klinis Unit Fungsi Paru University of Rome ldquoLa Sapienzardquo Italia
Abstrak Meskipun kor pulmonal merupakan penyebab utama kematian pada PPOK
lanjutan tidak ada konsensus yang jelas pada definisi tersebut Hipertensi pulmoner secara
tradisional dianggap sebagai mekanisme patogenetik yang mengarah ke disfungsi jantung
kanan pada PPOK dengan edema perifer yang biasanya menjadi dihubungkan dengan
kongesti vena sekunder untuk menaikkan tekanan pengisian ventrikel kanan Kerugian pada
kapiler paru dan remodeling vaskular secara sekunder terhadap penyebab hipoksia kronis
rokok asap rokok eksposisi dan inflamasi sistemik yang terkenal disebabkan oleh disfungsi
endotel dan hipertensi pulmonal pada pasien PPOK Mekanisme lain seperti vasokonstriksi
hipoksia dan kompresi kapiler alveolar pada paru yang hiperinflasi dapat berkontribusi pada
peningkatan tekanan arteri paru selama eksaserbasi PPOK olahraga dan tidur Dalam dekade
terakhir bagaimanapun juga peran tekanan paru yang berlebih dalam pengembangan gagal
jantung kanan telah diperdebatkan oleh kebanyakan orang karena kurangnya korelasi yang
jelas antara tekanan arteri pulmonalis dan adanya edema perifer dan atau tanda-tanda
hemodinamik gagal jantung kanan Selain itu adanya perubahan ginjal dan hormonal yang
berbeda menunjukkan bahwa pembentukan edema pada fase lanjutan dari PPOK sebagian
besar disebabkan oleh retensi natrium dan air yang disebabkan oleh asidosis hiperkapnia dan
diperparah oleh hipoksemia berat Penelitian selanjutnya adalah diperlukan untuk
memperjelas kemungkinan peran hipoksia intermiten yang ditimbulkan oleh latihan atau
tidur pada fungsi jantung kanan dan juga penanganan air dan natrium pada pasien PPOK
Kata kunci Hipertensi Pulmoner Remodeling vaskular hiperinflasi paru disfungsi jantung
kanan
1 PEMBUKAAN
Kor pulmonal merupakan komplikasi tahap akhir penyakit paru obstruktif kronis
(PPOK) yang merupakan penyebab kecacatan parah dan kematian Saat ini tidak ada
konsensus tentang definisi kor pulmonal mungkin karena pemahaman yang tidak lengkap
tentang mekanisme patogenetik yang bertanggung jawab untuk kondisi klinis ini Empat
puluh lima tahun lalu sebuah komite ahli dari WHO mendefinisikan kor pulmonal sebagai
hipertrofi ventrikel kanan akibat penyakit yang mempengaruhi fungsi dan atau struktur
paru-paru [1] Selanjutnya telah diusulkan untuk menggantikan istilah hipertrofi
dengan yang definisi yang berbeda tentang keterlibatan jantung kanan misalnya
perubahan dalam struktur dan fungsi ventrikel kanan [2]
Hipertensi pulmoner secara tradisional dianggap mekanisme patogenetik utama
disfungsi jantung kanan pada PPOK dan edema perifer telah dikaitkan ekstravasasi cairan
kapiler karena kongesti vena sekunder untuk transmisi hulu dari tekanan pengisian ventrikel
kanan Hal ini juga diketahui bagaimanapun juga bahwa beberapa pasien mungkin
mengalami edema perifer tanpa adanya perubahan hemodinamik yang signifikan dalam
kinerja jantung kanan lebih jauh lagi tingkat hipertensi pulmonal biasanya ringan pada
PPOK bahkan selama latihan tidur dan eksaserbasi penyakit dan tidak membenarkan
kehadiran edema perifer Hal ini juga telah diusulkan bahwa edema perifer pada PPOK
mungkin karena kelainan ginjal dan hormonal terkait dengan retensi natrium dan ekspansi
volume ekstra seluler Karena kurangnya korelasi erat antara disfungsi jantung kanan
(misalnya penurunan curah jantung) dan keparahan gejala klinis (misalnya dyspnea) dan
tanda-tanda (misalnya perifer edema) telah disarankan untuk memanfaatkan istilah kor
pulmonal untuk menentukan kondisi klinis yang ditandai dengan retensi air dan natrium
yang biasanya mempengaruhi pasien PPOK dengan kegagalan hiperkapnia pernafasan kronis
[3 4]
Tujuan dari review singkat ini adalah untuk membahas mekanisme yang terlibat
dalam patogenesis yang disebut kronis kor pulmonal (Tabel 1) Secara khusus bab ini akan
fokus pada mekanisme terpadu dan akuisisi patogenetik yang baru yang mungkin dapat
membantu untuk menjelaskan kemungkinan perbedaan antara pengukuran fisiologis obyektif
dan temuan klinis yang diamati dalam kor pulmonal
TABEL 1 Mekanisme Patogenetik Kor Pulmonal pada PPOK
Hipertensi Pulmoner Vasokontriksi hipoksia pulmoner
Remodeling endotel pulmoner
Hiperinflasi Paru
Desaturasi oksigen arteri selama latihan dan
tidur
Abnormalitas Ginjal dan Hormon Aktivasi Simpatis
Aktivasi sistem renin angiotensin aldosteron
Peningkatan level vasopresin
2 HIPERTENSI PULMONER PADA PPOK
Hipertensi pulmoner (HP) terkait dengan penyakit pernapasan kronis yang secara umum
didefinisikan sebagai rata-rata tekanan istirahat arteri pulmonalis (mPAP)gt 25 mmHg dan
atau gt 30 mmHg selama latihan [5] Pengukuran yang benar mPAP membutuhkan
kateterisasi jantung kanan yang tetap merupakan gold standard untuk diagnosis HP Pada
PPOK seperti pada penyakit pernapasan kronis lainnya hasil HP dari kenaikan resistensi
vaskuler paru (RVP) dalam adanya yang curah jantung jantung dan tekanan kapiler paru
normal baji (misalnya HP pra-kapiler) Insiden yang sebenarnya HP pada PPOK tidak
diketahui terutama karena kateterisasi jantung kanan belum dimanfaatkan secara sistematis
dalam spektrum yang luas dari PPOK Pada pasien dengan PPOK berat dan hipoksemia
istirahat ringan HP telah dilaporkan sampai dengan 20 dari kasus [6] Selain itu ketika
diukur pada saat istirahat HP umumnya ringan (misalnya lt35 mmHg) dan hanya sebagian
kecil pasien dengan PPOK berat menunjukkan tingkat istirahat mPAP untuk memenuhi
definisi moderat (mPAP lt45 mmHg) atau berat (mPAP 1048577 45 mmHg) HP [7] Pemberitahuan
prevalensi HP tersebut lebih tinggi pada pasien emfisema (pink puffers ) daripada bronkitis
kronis ( blue bloaters ) [8] Akhirnya mPAP telah dilaporkan untuk meningkatkan secara
bertahap selama perjalanan PPOK dengan tingkat kenaikan menengah 05 mmHg tahun
peningkatan mPAP terkait erat dengan memburuknya gas darah arteri [9]
Berbagai faktor dapat berkontribusi untuk pengembangan dan perawatan HP pada
PPOK vasokonstriksi hipoksia dan hipoksia yang diinduksi remodeling pembuluh pulmoner
telah dianggap sebagai faktor penyebab utama Pada tahun-tahun terakhir pemahaman yang
lebih baik dari kelainan sentral dan perifer yang terjadi pada PPOK menyebabkan
wawasan baru dalam patogenesis HP yang juga akan ditinjau dalam bab ini
21 Vasokontriksi Hipoksia Pulmoner
Dikenal sejak tahun 1946 penelitian Von Euler dan Liljestrand pada kucing
vasokonstriksi hipoksia pulmoner (VHP) telah diakui sebagai respon vasomotor adaptif
terhadap hipoksia alveolar yang menentukan redistribusi darah secara optimal yang segmen
paru yang berventilasi Dengan adanya hipoksia global seperti yang terjadi di dataran tinggi
atau pada pasien dengan sleep apnea VHP menyempitkan pre-kapiler pulmoner otot arteri
pulmonalis kecil seluruh sirkulasi paru dengan meningkatkan resistensi pembuluh darah
pulmoner (RPP) VHP ditimbulkan oleh hipoksia moderat dan hal itu mungkin meningkat
kan RPP menjadi 50-300 VHP mengembangkan respon terhadap hipoksia alveolar dalam
beberapa menit yang mencapai maksimum pada 15 menit VHP mungkin bertahan secara
kronis atau kembali normal saat kondisi normoxia dipulihkan [10] Mekanisme intraseluler
yang mendasari VHP sangat kompleks dan tidak sepenuhnya dipahami VHP merupakan
mekanisme intrinsik paru-paru dan intinya adalah dalam sel otot polos Teori terbaru untuk
VHP mengusulkan bahwa hipoksia yang diinduksi aktivasi sensor redoks mitokondria
dengan menghasilkan mediator difus seperti spesies oksigen reaktif menghambat aktivitas
jembatan tegangan saluran kalium Akibat depolarisasi sel-sel otot polos diikuti oleh
masuknya Ca2+ intraseluler dan akhirnya vasokonstriksi[10] VHP mungkin berkontribusi
terhadap munculnya tekanan pulmoner selama peristiwa hipoksia akut pada PPOK sebagai
penyakit eksaserbasi dan latihan dan atau tidur yang berhubungan dengan desaturasi
oksigen
Eksaserbasi akut adalah kejadian umum pada PPOK dan frekuensinya berkorelasi
dengan keparahan penyakit Episode ini biasanya terkait dengan memburuknya hipoksemia
dan hiperkapnia terkait dengan peningkatan mPAP [2] Selain itu dalam PPOK berat
eksaserbasi akut berhubungan dengan onset atau memburuknya edema perifer Banyak faktor
yang mungkin terlibat dalam memburuknya HP selama eksaserbasi PPOK hipoksemia
alveolar dapat menyebabkan vasokonstriksi paru akut dan hiperkapnia dapat berkontribusi
pada peningkatan RPP dengan meningkatkan tonus arteri pulmonalis
VHP juga dapat menjadi penyebab kenaikan mPAP yang diamati pada pasien PPOK
selama tidur REM ketika jatuh pada saturasi oksigen lebih jelas Bahkan tidur terkait
desaturasi oksigen pada PPOK tidak selalu disebabkan oleh apnea (kecuali bila PPOK
dikaitkan dengan sleep apnea syndrome) tetapi dengan tidur terkait memburuknya ventilasi
alveolar dan atau ventilasi-perfusi Desaturasi oksigen yang lebih besar merupakan hal yang
lebih parah adalah pada peningkatan di HP dengan mPAP yang dapat meningkatkan
sebanyak 10 mmHg dari itu nilai dasar [11]
22 Remodeling Pulmoner Vaskular
Pasien dengan stadium akhir PPOK menunjukkan perubahan mencolok dalam otot
arteri pulmonal dan pembuluh pre-kapiler yang menjelaskan peningkatan ireversibel pada
RPP Pada pasien ini studi post-mortem telah mengungkapkan deposisi otot membujur dan
fibrosis dari intima dari otot arteri pulmoner dengan perkembangan lapisan medial melingkar
otot polos yang dibatasi oleh lamina elastis baru di arteriol
[1213] (Gambar 1)
Gambar 1 Remodeling vaskular pulmoner pada PPOK Catat penebalan intima prominen dan
penyempitan lumen (diambil dari ref [10] dengan ijin)
Remodeling vaskular pulmoner pada penyakit pernapasan biasanya berkembang
dengan adanya hipoksia kronis (PaO2 lt 55-60 mmHg) Dalam studi in vitro telah
menunjukkan bahwa hipoksia menginduksi perubahan dalam produksi dan pelepasan
substansi endotheliumderived vasoaktif proliferasi sel tersebut pada dinding pembuluh darah
serta sintesis peningkatan matriks protein ekstra-seluler Hipoksia yang diinduksi
muskuluarisasi dari prekapiler arteriol yang awalnya dikaitkan dengan VHP persisten
alternatifnya hal tersebut bisa diturunkan dengan transdiferensiasi tersebut sel endotel
menjadi otot polos sebagai respon adaptif dari stres arteri pra-kapiler [14]
Menariknya kelainan pembuluh darah pulmoner terutama yang terdiri dari penebalan
intima dari otot arteri pulmoner oleh proliferasi sel-sel otot polos dan deposisi dari kedua
serat elastis dan kolagen juga telah didokumentasikan pada pasien non-hipoksemia ringan
dan pada perokok dengan fungsi paru-paru normal [15] Pengamatan ini menunjukkan itu
bersama-sama dengan hipoksemia kronis faktor lain bisa memiliki peran dalam patogenesis
HP dan disfungsi endotel dalam fase awal dari PPOK
Penelitian terbaru menunjukkan bahwa kerugian fungsi endotel mungkin timbul dari
perubahan dalam ekspresi dan pelepasan mediator vasoaktif yang penting seperti nitrat
oksida (NO) dan endotelin (ET-1) NO merupakan vasodilator kuat dengan sifat
antiproliferatif yang disintesis oleh NO sintetase (NOS) yang diungkapkan secara konstitutif
pada sel endotel Ekspresi NOS berkurang di arteri pulmonalis Pasien PPOK dengan HP dan
mungkin adalah penyebab dari peningkatan regulasi tersebut dan ekspresi gen yang
mengkodifikasi pada beberapa faktor pertumbuhan seperti faktor pertumbuhan endotel
vaskular [16] Mekanisme ini dapat menyebabkan proliferasi sel dan remodeling pembuluh
arteri pulmonalis ET-1 adalah vasokonstriktor kuat dilepaskan oleh sel endotel yang juga
memberikan suatu efek mitogenik pada otot polos arteri sel ekspresi ET-1 meningkat pada
arteri pulmonalis dari pasien dengan HP hal tersebut menunjukkan peran yang mungkin dari
mediator ini untuk berkontribusi dalam remodeling vaskuler [17]
Adanya perubahan struktural dalam arteri pulmonalis pada perokok dengan fungsi
paru-paru normal dibandingkan dengan non-perokok menunjukkan efek langsung dari
produk rokok-rokok pada struktur pembuluh darab di sisi lain juga diketahui bahwa rokok-
rokok menginduksi disfungsi endotel pada arteri koroner dan sistemik Eksposisi aktif dan
pasif pada asap tembakau mungkin bertindak dengan mengurangi aktivitas dan ekspresi NOS
pada arteri pulmonalis dengan gangguan yang dihasilkan dari fungsi endotel dan remodeling
pembuluh pre-kapiler [18] Merokok juga bisa menyebabkan perubahan inflamasi di arteri
pulmonalis Baru-baru ini peningkatan jumlah infiltrasi sel-sel inflamasi tersebut pada
adventisia arteri pulmonalis pasien dengan PPOK telah dijelaskan infiltrat inflamasi lokal
seperti didasari oleh diaktifkannya T-limfosit dengan dominasi suatu dari CD8 + bagian [19]
Sel-sel inflamasi dengan melepaskan sitokin dan faktor pertumbuhan yang dapat
menargetkan sel endotel dapat memberikan kontribusi untuk pengembangan struktural dan
kelainan fungsional dari dinding pembuluh darah [20]
Ada semakin banyak bukti peradangan sistemik pada PPOK seperti yang ditunjukkan
oleh peningkatan tingkat sistemik protein inflamasi seperti protein C-reaktif (CRP) tumor
necrosis factor α (TNF - α) dan interleukin 6 (IL-6) pada pasien ini [21] Baru-baru ini
menunjukkan bahwa pasien PPOK dengan hipertensi pulmonal memiliki serum CRP dan
TNF- α yang lebih tinggi tingkat dibandingkan dengan mereka yang normal tingkat
mPAPnya hal tersebut menunjukkan peran yang mungkin dari peradangan sistemik dalam
patogenesis hipertensi pulmoner [22] Dalam sel endotel manusia CRP mengurangi ekspresi
endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

hipertrofi ventrikel kanan akibat penyakit yang mempengaruhi fungsi dan atau struktur
paru-paru [1] Selanjutnya telah diusulkan untuk menggantikan istilah hipertrofi
dengan yang definisi yang berbeda tentang keterlibatan jantung kanan misalnya
perubahan dalam struktur dan fungsi ventrikel kanan [2]
Hipertensi pulmoner secara tradisional dianggap mekanisme patogenetik utama
disfungsi jantung kanan pada PPOK dan edema perifer telah dikaitkan ekstravasasi cairan
kapiler karena kongesti vena sekunder untuk transmisi hulu dari tekanan pengisian ventrikel
kanan Hal ini juga diketahui bagaimanapun juga bahwa beberapa pasien mungkin
mengalami edema perifer tanpa adanya perubahan hemodinamik yang signifikan dalam
kinerja jantung kanan lebih jauh lagi tingkat hipertensi pulmonal biasanya ringan pada
PPOK bahkan selama latihan tidur dan eksaserbasi penyakit dan tidak membenarkan
kehadiran edema perifer Hal ini juga telah diusulkan bahwa edema perifer pada PPOK
mungkin karena kelainan ginjal dan hormonal terkait dengan retensi natrium dan ekspansi
volume ekstra seluler Karena kurangnya korelasi erat antara disfungsi jantung kanan
(misalnya penurunan curah jantung) dan keparahan gejala klinis (misalnya dyspnea) dan
tanda-tanda (misalnya perifer edema) telah disarankan untuk memanfaatkan istilah kor
pulmonal untuk menentukan kondisi klinis yang ditandai dengan retensi air dan natrium
yang biasanya mempengaruhi pasien PPOK dengan kegagalan hiperkapnia pernafasan kronis
[3 4]
Tujuan dari review singkat ini adalah untuk membahas mekanisme yang terlibat
dalam patogenesis yang disebut kronis kor pulmonal (Tabel 1) Secara khusus bab ini akan
fokus pada mekanisme terpadu dan akuisisi patogenetik yang baru yang mungkin dapat
membantu untuk menjelaskan kemungkinan perbedaan antara pengukuran fisiologis obyektif
dan temuan klinis yang diamati dalam kor pulmonal
TABEL 1 Mekanisme Patogenetik Kor Pulmonal pada PPOK
Hipertensi Pulmoner Vasokontriksi hipoksia pulmoner
Remodeling endotel pulmoner
Hiperinflasi Paru
Desaturasi oksigen arteri selama latihan dan
tidur
Abnormalitas Ginjal dan Hormon Aktivasi Simpatis
Aktivasi sistem renin angiotensin aldosteron
Peningkatan level vasopresin
2 HIPERTENSI PULMONER PADA PPOK
Hipertensi pulmoner (HP) terkait dengan penyakit pernapasan kronis yang secara umum
didefinisikan sebagai rata-rata tekanan istirahat arteri pulmonalis (mPAP)gt 25 mmHg dan
atau gt 30 mmHg selama latihan [5] Pengukuran yang benar mPAP membutuhkan
kateterisasi jantung kanan yang tetap merupakan gold standard untuk diagnosis HP Pada
PPOK seperti pada penyakit pernapasan kronis lainnya hasil HP dari kenaikan resistensi
vaskuler paru (RVP) dalam adanya yang curah jantung jantung dan tekanan kapiler paru
normal baji (misalnya HP pra-kapiler) Insiden yang sebenarnya HP pada PPOK tidak
diketahui terutama karena kateterisasi jantung kanan belum dimanfaatkan secara sistematis
dalam spektrum yang luas dari PPOK Pada pasien dengan PPOK berat dan hipoksemia
istirahat ringan HP telah dilaporkan sampai dengan 20 dari kasus [6] Selain itu ketika
diukur pada saat istirahat HP umumnya ringan (misalnya lt35 mmHg) dan hanya sebagian
kecil pasien dengan PPOK berat menunjukkan tingkat istirahat mPAP untuk memenuhi
definisi moderat (mPAP lt45 mmHg) atau berat (mPAP 1048577 45 mmHg) HP [7] Pemberitahuan
prevalensi HP tersebut lebih tinggi pada pasien emfisema (pink puffers ) daripada bronkitis
kronis ( blue bloaters ) [8] Akhirnya mPAP telah dilaporkan untuk meningkatkan secara
bertahap selama perjalanan PPOK dengan tingkat kenaikan menengah 05 mmHg tahun
peningkatan mPAP terkait erat dengan memburuknya gas darah arteri [9]
Berbagai faktor dapat berkontribusi untuk pengembangan dan perawatan HP pada
PPOK vasokonstriksi hipoksia dan hipoksia yang diinduksi remodeling pembuluh pulmoner
telah dianggap sebagai faktor penyebab utama Pada tahun-tahun terakhir pemahaman yang
lebih baik dari kelainan sentral dan perifer yang terjadi pada PPOK menyebabkan
wawasan baru dalam patogenesis HP yang juga akan ditinjau dalam bab ini
21 Vasokontriksi Hipoksia Pulmoner
Dikenal sejak tahun 1946 penelitian Von Euler dan Liljestrand pada kucing
vasokonstriksi hipoksia pulmoner (VHP) telah diakui sebagai respon vasomotor adaptif
terhadap hipoksia alveolar yang menentukan redistribusi darah secara optimal yang segmen
paru yang berventilasi Dengan adanya hipoksia global seperti yang terjadi di dataran tinggi
atau pada pasien dengan sleep apnea VHP menyempitkan pre-kapiler pulmoner otot arteri
pulmonalis kecil seluruh sirkulasi paru dengan meningkatkan resistensi pembuluh darah
pulmoner (RPP) VHP ditimbulkan oleh hipoksia moderat dan hal itu mungkin meningkat
kan RPP menjadi 50-300 VHP mengembangkan respon terhadap hipoksia alveolar dalam
beberapa menit yang mencapai maksimum pada 15 menit VHP mungkin bertahan secara
kronis atau kembali normal saat kondisi normoxia dipulihkan [10] Mekanisme intraseluler
yang mendasari VHP sangat kompleks dan tidak sepenuhnya dipahami VHP merupakan
mekanisme intrinsik paru-paru dan intinya adalah dalam sel otot polos Teori terbaru untuk
VHP mengusulkan bahwa hipoksia yang diinduksi aktivasi sensor redoks mitokondria
dengan menghasilkan mediator difus seperti spesies oksigen reaktif menghambat aktivitas
jembatan tegangan saluran kalium Akibat depolarisasi sel-sel otot polos diikuti oleh
masuknya Ca2+ intraseluler dan akhirnya vasokonstriksi[10] VHP mungkin berkontribusi
terhadap munculnya tekanan pulmoner selama peristiwa hipoksia akut pada PPOK sebagai
penyakit eksaserbasi dan latihan dan atau tidur yang berhubungan dengan desaturasi
oksigen
Eksaserbasi akut adalah kejadian umum pada PPOK dan frekuensinya berkorelasi
dengan keparahan penyakit Episode ini biasanya terkait dengan memburuknya hipoksemia
dan hiperkapnia terkait dengan peningkatan mPAP [2] Selain itu dalam PPOK berat
eksaserbasi akut berhubungan dengan onset atau memburuknya edema perifer Banyak faktor
yang mungkin terlibat dalam memburuknya HP selama eksaserbasi PPOK hipoksemia
alveolar dapat menyebabkan vasokonstriksi paru akut dan hiperkapnia dapat berkontribusi
pada peningkatan RPP dengan meningkatkan tonus arteri pulmonalis
VHP juga dapat menjadi penyebab kenaikan mPAP yang diamati pada pasien PPOK
selama tidur REM ketika jatuh pada saturasi oksigen lebih jelas Bahkan tidur terkait
desaturasi oksigen pada PPOK tidak selalu disebabkan oleh apnea (kecuali bila PPOK
dikaitkan dengan sleep apnea syndrome) tetapi dengan tidur terkait memburuknya ventilasi
alveolar dan atau ventilasi-perfusi Desaturasi oksigen yang lebih besar merupakan hal yang
lebih parah adalah pada peningkatan di HP dengan mPAP yang dapat meningkatkan
sebanyak 10 mmHg dari itu nilai dasar [11]
22 Remodeling Pulmoner Vaskular
Pasien dengan stadium akhir PPOK menunjukkan perubahan mencolok dalam otot
arteri pulmonal dan pembuluh pre-kapiler yang menjelaskan peningkatan ireversibel pada
RPP Pada pasien ini studi post-mortem telah mengungkapkan deposisi otot membujur dan
fibrosis dari intima dari otot arteri pulmoner dengan perkembangan lapisan medial melingkar
otot polos yang dibatasi oleh lamina elastis baru di arteriol
[1213] (Gambar 1)
Gambar 1 Remodeling vaskular pulmoner pada PPOK Catat penebalan intima prominen dan
penyempitan lumen (diambil dari ref [10] dengan ijin)
Remodeling vaskular pulmoner pada penyakit pernapasan biasanya berkembang
dengan adanya hipoksia kronis (PaO2 lt 55-60 mmHg) Dalam studi in vitro telah
menunjukkan bahwa hipoksia menginduksi perubahan dalam produksi dan pelepasan
substansi endotheliumderived vasoaktif proliferasi sel tersebut pada dinding pembuluh darah
serta sintesis peningkatan matriks protein ekstra-seluler Hipoksia yang diinduksi
muskuluarisasi dari prekapiler arteriol yang awalnya dikaitkan dengan VHP persisten
alternatifnya hal tersebut bisa diturunkan dengan transdiferensiasi tersebut sel endotel
menjadi otot polos sebagai respon adaptif dari stres arteri pra-kapiler [14]
Menariknya kelainan pembuluh darah pulmoner terutama yang terdiri dari penebalan
intima dari otot arteri pulmoner oleh proliferasi sel-sel otot polos dan deposisi dari kedua
serat elastis dan kolagen juga telah didokumentasikan pada pasien non-hipoksemia ringan
dan pada perokok dengan fungsi paru-paru normal [15] Pengamatan ini menunjukkan itu
bersama-sama dengan hipoksemia kronis faktor lain bisa memiliki peran dalam patogenesis
HP dan disfungsi endotel dalam fase awal dari PPOK
Penelitian terbaru menunjukkan bahwa kerugian fungsi endotel mungkin timbul dari
perubahan dalam ekspresi dan pelepasan mediator vasoaktif yang penting seperti nitrat
oksida (NO) dan endotelin (ET-1) NO merupakan vasodilator kuat dengan sifat
antiproliferatif yang disintesis oleh NO sintetase (NOS) yang diungkapkan secara konstitutif
pada sel endotel Ekspresi NOS berkurang di arteri pulmonalis Pasien PPOK dengan HP dan
mungkin adalah penyebab dari peningkatan regulasi tersebut dan ekspresi gen yang
mengkodifikasi pada beberapa faktor pertumbuhan seperti faktor pertumbuhan endotel
vaskular [16] Mekanisme ini dapat menyebabkan proliferasi sel dan remodeling pembuluh
arteri pulmonalis ET-1 adalah vasokonstriktor kuat dilepaskan oleh sel endotel yang juga
memberikan suatu efek mitogenik pada otot polos arteri sel ekspresi ET-1 meningkat pada
arteri pulmonalis dari pasien dengan HP hal tersebut menunjukkan peran yang mungkin dari
mediator ini untuk berkontribusi dalam remodeling vaskuler [17]
Adanya perubahan struktural dalam arteri pulmonalis pada perokok dengan fungsi
paru-paru normal dibandingkan dengan non-perokok menunjukkan efek langsung dari
produk rokok-rokok pada struktur pembuluh darab di sisi lain juga diketahui bahwa rokok-
rokok menginduksi disfungsi endotel pada arteri koroner dan sistemik Eksposisi aktif dan
pasif pada asap tembakau mungkin bertindak dengan mengurangi aktivitas dan ekspresi NOS
pada arteri pulmonalis dengan gangguan yang dihasilkan dari fungsi endotel dan remodeling
pembuluh pre-kapiler [18] Merokok juga bisa menyebabkan perubahan inflamasi di arteri
pulmonalis Baru-baru ini peningkatan jumlah infiltrasi sel-sel inflamasi tersebut pada
adventisia arteri pulmonalis pasien dengan PPOK telah dijelaskan infiltrat inflamasi lokal
seperti didasari oleh diaktifkannya T-limfosit dengan dominasi suatu dari CD8 + bagian [19]
Sel-sel inflamasi dengan melepaskan sitokin dan faktor pertumbuhan yang dapat
menargetkan sel endotel dapat memberikan kontribusi untuk pengembangan struktural dan
kelainan fungsional dari dinding pembuluh darah [20]
Ada semakin banyak bukti peradangan sistemik pada PPOK seperti yang ditunjukkan
oleh peningkatan tingkat sistemik protein inflamasi seperti protein C-reaktif (CRP) tumor
necrosis factor α (TNF - α) dan interleukin 6 (IL-6) pada pasien ini [21] Baru-baru ini
menunjukkan bahwa pasien PPOK dengan hipertensi pulmonal memiliki serum CRP dan
TNF- α yang lebih tinggi tingkat dibandingkan dengan mereka yang normal tingkat
mPAPnya hal tersebut menunjukkan peran yang mungkin dari peradangan sistemik dalam
patogenesis hipertensi pulmoner [22] Dalam sel endotel manusia CRP mengurangi ekspresi
endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

Aktivasi sistem renin angiotensin aldosteron
Peningkatan level vasopresin
2 HIPERTENSI PULMONER PADA PPOK
Hipertensi pulmoner (HP) terkait dengan penyakit pernapasan kronis yang secara umum
didefinisikan sebagai rata-rata tekanan istirahat arteri pulmonalis (mPAP)gt 25 mmHg dan
atau gt 30 mmHg selama latihan [5] Pengukuran yang benar mPAP membutuhkan
kateterisasi jantung kanan yang tetap merupakan gold standard untuk diagnosis HP Pada
PPOK seperti pada penyakit pernapasan kronis lainnya hasil HP dari kenaikan resistensi
vaskuler paru (RVP) dalam adanya yang curah jantung jantung dan tekanan kapiler paru
normal baji (misalnya HP pra-kapiler) Insiden yang sebenarnya HP pada PPOK tidak
diketahui terutama karena kateterisasi jantung kanan belum dimanfaatkan secara sistematis
dalam spektrum yang luas dari PPOK Pada pasien dengan PPOK berat dan hipoksemia
istirahat ringan HP telah dilaporkan sampai dengan 20 dari kasus [6] Selain itu ketika
diukur pada saat istirahat HP umumnya ringan (misalnya lt35 mmHg) dan hanya sebagian
kecil pasien dengan PPOK berat menunjukkan tingkat istirahat mPAP untuk memenuhi
definisi moderat (mPAP lt45 mmHg) atau berat (mPAP 1048577 45 mmHg) HP [7] Pemberitahuan
prevalensi HP tersebut lebih tinggi pada pasien emfisema (pink puffers ) daripada bronkitis
kronis ( blue bloaters ) [8] Akhirnya mPAP telah dilaporkan untuk meningkatkan secara
bertahap selama perjalanan PPOK dengan tingkat kenaikan menengah 05 mmHg tahun
peningkatan mPAP terkait erat dengan memburuknya gas darah arteri [9]
Berbagai faktor dapat berkontribusi untuk pengembangan dan perawatan HP pada
PPOK vasokonstriksi hipoksia dan hipoksia yang diinduksi remodeling pembuluh pulmoner
telah dianggap sebagai faktor penyebab utama Pada tahun-tahun terakhir pemahaman yang
lebih baik dari kelainan sentral dan perifer yang terjadi pada PPOK menyebabkan
wawasan baru dalam patogenesis HP yang juga akan ditinjau dalam bab ini
21 Vasokontriksi Hipoksia Pulmoner
Dikenal sejak tahun 1946 penelitian Von Euler dan Liljestrand pada kucing
vasokonstriksi hipoksia pulmoner (VHP) telah diakui sebagai respon vasomotor adaptif
terhadap hipoksia alveolar yang menentukan redistribusi darah secara optimal yang segmen
paru yang berventilasi Dengan adanya hipoksia global seperti yang terjadi di dataran tinggi
atau pada pasien dengan sleep apnea VHP menyempitkan pre-kapiler pulmoner otot arteri
pulmonalis kecil seluruh sirkulasi paru dengan meningkatkan resistensi pembuluh darah
pulmoner (RPP) VHP ditimbulkan oleh hipoksia moderat dan hal itu mungkin meningkat
kan RPP menjadi 50-300 VHP mengembangkan respon terhadap hipoksia alveolar dalam
beberapa menit yang mencapai maksimum pada 15 menit VHP mungkin bertahan secara
kronis atau kembali normal saat kondisi normoxia dipulihkan [10] Mekanisme intraseluler
yang mendasari VHP sangat kompleks dan tidak sepenuhnya dipahami VHP merupakan
mekanisme intrinsik paru-paru dan intinya adalah dalam sel otot polos Teori terbaru untuk
VHP mengusulkan bahwa hipoksia yang diinduksi aktivasi sensor redoks mitokondria
dengan menghasilkan mediator difus seperti spesies oksigen reaktif menghambat aktivitas
jembatan tegangan saluran kalium Akibat depolarisasi sel-sel otot polos diikuti oleh
masuknya Ca2+ intraseluler dan akhirnya vasokonstriksi[10] VHP mungkin berkontribusi
terhadap munculnya tekanan pulmoner selama peristiwa hipoksia akut pada PPOK sebagai
penyakit eksaserbasi dan latihan dan atau tidur yang berhubungan dengan desaturasi
oksigen
Eksaserbasi akut adalah kejadian umum pada PPOK dan frekuensinya berkorelasi
dengan keparahan penyakit Episode ini biasanya terkait dengan memburuknya hipoksemia
dan hiperkapnia terkait dengan peningkatan mPAP [2] Selain itu dalam PPOK berat
eksaserbasi akut berhubungan dengan onset atau memburuknya edema perifer Banyak faktor
yang mungkin terlibat dalam memburuknya HP selama eksaserbasi PPOK hipoksemia
alveolar dapat menyebabkan vasokonstriksi paru akut dan hiperkapnia dapat berkontribusi
pada peningkatan RPP dengan meningkatkan tonus arteri pulmonalis
VHP juga dapat menjadi penyebab kenaikan mPAP yang diamati pada pasien PPOK
selama tidur REM ketika jatuh pada saturasi oksigen lebih jelas Bahkan tidur terkait
desaturasi oksigen pada PPOK tidak selalu disebabkan oleh apnea (kecuali bila PPOK
dikaitkan dengan sleep apnea syndrome) tetapi dengan tidur terkait memburuknya ventilasi
alveolar dan atau ventilasi-perfusi Desaturasi oksigen yang lebih besar merupakan hal yang
lebih parah adalah pada peningkatan di HP dengan mPAP yang dapat meningkatkan
sebanyak 10 mmHg dari itu nilai dasar [11]
22 Remodeling Pulmoner Vaskular
Pasien dengan stadium akhir PPOK menunjukkan perubahan mencolok dalam otot
arteri pulmonal dan pembuluh pre-kapiler yang menjelaskan peningkatan ireversibel pada
RPP Pada pasien ini studi post-mortem telah mengungkapkan deposisi otot membujur dan
fibrosis dari intima dari otot arteri pulmoner dengan perkembangan lapisan medial melingkar
otot polos yang dibatasi oleh lamina elastis baru di arteriol
[1213] (Gambar 1)
Gambar 1 Remodeling vaskular pulmoner pada PPOK Catat penebalan intima prominen dan
penyempitan lumen (diambil dari ref [10] dengan ijin)
Remodeling vaskular pulmoner pada penyakit pernapasan biasanya berkembang
dengan adanya hipoksia kronis (PaO2 lt 55-60 mmHg) Dalam studi in vitro telah
menunjukkan bahwa hipoksia menginduksi perubahan dalam produksi dan pelepasan
substansi endotheliumderived vasoaktif proliferasi sel tersebut pada dinding pembuluh darah
serta sintesis peningkatan matriks protein ekstra-seluler Hipoksia yang diinduksi
muskuluarisasi dari prekapiler arteriol yang awalnya dikaitkan dengan VHP persisten
alternatifnya hal tersebut bisa diturunkan dengan transdiferensiasi tersebut sel endotel
menjadi otot polos sebagai respon adaptif dari stres arteri pra-kapiler [14]
Menariknya kelainan pembuluh darah pulmoner terutama yang terdiri dari penebalan
intima dari otot arteri pulmoner oleh proliferasi sel-sel otot polos dan deposisi dari kedua
serat elastis dan kolagen juga telah didokumentasikan pada pasien non-hipoksemia ringan
dan pada perokok dengan fungsi paru-paru normal [15] Pengamatan ini menunjukkan itu
bersama-sama dengan hipoksemia kronis faktor lain bisa memiliki peran dalam patogenesis
HP dan disfungsi endotel dalam fase awal dari PPOK
Penelitian terbaru menunjukkan bahwa kerugian fungsi endotel mungkin timbul dari
perubahan dalam ekspresi dan pelepasan mediator vasoaktif yang penting seperti nitrat
oksida (NO) dan endotelin (ET-1) NO merupakan vasodilator kuat dengan sifat
antiproliferatif yang disintesis oleh NO sintetase (NOS) yang diungkapkan secara konstitutif
pada sel endotel Ekspresi NOS berkurang di arteri pulmonalis Pasien PPOK dengan HP dan
mungkin adalah penyebab dari peningkatan regulasi tersebut dan ekspresi gen yang
mengkodifikasi pada beberapa faktor pertumbuhan seperti faktor pertumbuhan endotel
vaskular [16] Mekanisme ini dapat menyebabkan proliferasi sel dan remodeling pembuluh
arteri pulmonalis ET-1 adalah vasokonstriktor kuat dilepaskan oleh sel endotel yang juga
memberikan suatu efek mitogenik pada otot polos arteri sel ekspresi ET-1 meningkat pada
arteri pulmonalis dari pasien dengan HP hal tersebut menunjukkan peran yang mungkin dari
mediator ini untuk berkontribusi dalam remodeling vaskuler [17]
Adanya perubahan struktural dalam arteri pulmonalis pada perokok dengan fungsi
paru-paru normal dibandingkan dengan non-perokok menunjukkan efek langsung dari
produk rokok-rokok pada struktur pembuluh darab di sisi lain juga diketahui bahwa rokok-
rokok menginduksi disfungsi endotel pada arteri koroner dan sistemik Eksposisi aktif dan
pasif pada asap tembakau mungkin bertindak dengan mengurangi aktivitas dan ekspresi NOS
pada arteri pulmonalis dengan gangguan yang dihasilkan dari fungsi endotel dan remodeling
pembuluh pre-kapiler [18] Merokok juga bisa menyebabkan perubahan inflamasi di arteri
pulmonalis Baru-baru ini peningkatan jumlah infiltrasi sel-sel inflamasi tersebut pada
adventisia arteri pulmonalis pasien dengan PPOK telah dijelaskan infiltrat inflamasi lokal
seperti didasari oleh diaktifkannya T-limfosit dengan dominasi suatu dari CD8 + bagian [19]
Sel-sel inflamasi dengan melepaskan sitokin dan faktor pertumbuhan yang dapat
menargetkan sel endotel dapat memberikan kontribusi untuk pengembangan struktural dan
kelainan fungsional dari dinding pembuluh darah [20]
Ada semakin banyak bukti peradangan sistemik pada PPOK seperti yang ditunjukkan
oleh peningkatan tingkat sistemik protein inflamasi seperti protein C-reaktif (CRP) tumor
necrosis factor α (TNF - α) dan interleukin 6 (IL-6) pada pasien ini [21] Baru-baru ini
menunjukkan bahwa pasien PPOK dengan hipertensi pulmonal memiliki serum CRP dan
TNF- α yang lebih tinggi tingkat dibandingkan dengan mereka yang normal tingkat
mPAPnya hal tersebut menunjukkan peran yang mungkin dari peradangan sistemik dalam
patogenesis hipertensi pulmoner [22] Dalam sel endotel manusia CRP mengurangi ekspresi
endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

atau pada pasien dengan sleep apnea VHP menyempitkan pre-kapiler pulmoner otot arteri
pulmonalis kecil seluruh sirkulasi paru dengan meningkatkan resistensi pembuluh darah
pulmoner (RPP) VHP ditimbulkan oleh hipoksia moderat dan hal itu mungkin meningkat
kan RPP menjadi 50-300 VHP mengembangkan respon terhadap hipoksia alveolar dalam
beberapa menit yang mencapai maksimum pada 15 menit VHP mungkin bertahan secara
kronis atau kembali normal saat kondisi normoxia dipulihkan [10] Mekanisme intraseluler
yang mendasari VHP sangat kompleks dan tidak sepenuhnya dipahami VHP merupakan
mekanisme intrinsik paru-paru dan intinya adalah dalam sel otot polos Teori terbaru untuk
VHP mengusulkan bahwa hipoksia yang diinduksi aktivasi sensor redoks mitokondria
dengan menghasilkan mediator difus seperti spesies oksigen reaktif menghambat aktivitas
jembatan tegangan saluran kalium Akibat depolarisasi sel-sel otot polos diikuti oleh
masuknya Ca2+ intraseluler dan akhirnya vasokonstriksi[10] VHP mungkin berkontribusi
terhadap munculnya tekanan pulmoner selama peristiwa hipoksia akut pada PPOK sebagai
penyakit eksaserbasi dan latihan dan atau tidur yang berhubungan dengan desaturasi
oksigen
Eksaserbasi akut adalah kejadian umum pada PPOK dan frekuensinya berkorelasi
dengan keparahan penyakit Episode ini biasanya terkait dengan memburuknya hipoksemia
dan hiperkapnia terkait dengan peningkatan mPAP [2] Selain itu dalam PPOK berat
eksaserbasi akut berhubungan dengan onset atau memburuknya edema perifer Banyak faktor
yang mungkin terlibat dalam memburuknya HP selama eksaserbasi PPOK hipoksemia
alveolar dapat menyebabkan vasokonstriksi paru akut dan hiperkapnia dapat berkontribusi
pada peningkatan RPP dengan meningkatkan tonus arteri pulmonalis
VHP juga dapat menjadi penyebab kenaikan mPAP yang diamati pada pasien PPOK
selama tidur REM ketika jatuh pada saturasi oksigen lebih jelas Bahkan tidur terkait
desaturasi oksigen pada PPOK tidak selalu disebabkan oleh apnea (kecuali bila PPOK
dikaitkan dengan sleep apnea syndrome) tetapi dengan tidur terkait memburuknya ventilasi
alveolar dan atau ventilasi-perfusi Desaturasi oksigen yang lebih besar merupakan hal yang
lebih parah adalah pada peningkatan di HP dengan mPAP yang dapat meningkatkan
sebanyak 10 mmHg dari itu nilai dasar [11]
22 Remodeling Pulmoner Vaskular
Pasien dengan stadium akhir PPOK menunjukkan perubahan mencolok dalam otot
arteri pulmonal dan pembuluh pre-kapiler yang menjelaskan peningkatan ireversibel pada
RPP Pada pasien ini studi post-mortem telah mengungkapkan deposisi otot membujur dan
fibrosis dari intima dari otot arteri pulmoner dengan perkembangan lapisan medial melingkar
otot polos yang dibatasi oleh lamina elastis baru di arteriol
[1213] (Gambar 1)
Gambar 1 Remodeling vaskular pulmoner pada PPOK Catat penebalan intima prominen dan
penyempitan lumen (diambil dari ref [10] dengan ijin)
Remodeling vaskular pulmoner pada penyakit pernapasan biasanya berkembang
dengan adanya hipoksia kronis (PaO2 lt 55-60 mmHg) Dalam studi in vitro telah
menunjukkan bahwa hipoksia menginduksi perubahan dalam produksi dan pelepasan
substansi endotheliumderived vasoaktif proliferasi sel tersebut pada dinding pembuluh darah
serta sintesis peningkatan matriks protein ekstra-seluler Hipoksia yang diinduksi
muskuluarisasi dari prekapiler arteriol yang awalnya dikaitkan dengan VHP persisten
alternatifnya hal tersebut bisa diturunkan dengan transdiferensiasi tersebut sel endotel
menjadi otot polos sebagai respon adaptif dari stres arteri pra-kapiler [14]
Menariknya kelainan pembuluh darah pulmoner terutama yang terdiri dari penebalan
intima dari otot arteri pulmoner oleh proliferasi sel-sel otot polos dan deposisi dari kedua
serat elastis dan kolagen juga telah didokumentasikan pada pasien non-hipoksemia ringan
dan pada perokok dengan fungsi paru-paru normal [15] Pengamatan ini menunjukkan itu
bersama-sama dengan hipoksemia kronis faktor lain bisa memiliki peran dalam patogenesis
HP dan disfungsi endotel dalam fase awal dari PPOK
Penelitian terbaru menunjukkan bahwa kerugian fungsi endotel mungkin timbul dari
perubahan dalam ekspresi dan pelepasan mediator vasoaktif yang penting seperti nitrat
oksida (NO) dan endotelin (ET-1) NO merupakan vasodilator kuat dengan sifat
antiproliferatif yang disintesis oleh NO sintetase (NOS) yang diungkapkan secara konstitutif
pada sel endotel Ekspresi NOS berkurang di arteri pulmonalis Pasien PPOK dengan HP dan
mungkin adalah penyebab dari peningkatan regulasi tersebut dan ekspresi gen yang
mengkodifikasi pada beberapa faktor pertumbuhan seperti faktor pertumbuhan endotel
vaskular [16] Mekanisme ini dapat menyebabkan proliferasi sel dan remodeling pembuluh
arteri pulmonalis ET-1 adalah vasokonstriktor kuat dilepaskan oleh sel endotel yang juga
memberikan suatu efek mitogenik pada otot polos arteri sel ekspresi ET-1 meningkat pada
arteri pulmonalis dari pasien dengan HP hal tersebut menunjukkan peran yang mungkin dari
mediator ini untuk berkontribusi dalam remodeling vaskuler [17]
Adanya perubahan struktural dalam arteri pulmonalis pada perokok dengan fungsi
paru-paru normal dibandingkan dengan non-perokok menunjukkan efek langsung dari
produk rokok-rokok pada struktur pembuluh darab di sisi lain juga diketahui bahwa rokok-
rokok menginduksi disfungsi endotel pada arteri koroner dan sistemik Eksposisi aktif dan
pasif pada asap tembakau mungkin bertindak dengan mengurangi aktivitas dan ekspresi NOS
pada arteri pulmonalis dengan gangguan yang dihasilkan dari fungsi endotel dan remodeling
pembuluh pre-kapiler [18] Merokok juga bisa menyebabkan perubahan inflamasi di arteri
pulmonalis Baru-baru ini peningkatan jumlah infiltrasi sel-sel inflamasi tersebut pada
adventisia arteri pulmonalis pasien dengan PPOK telah dijelaskan infiltrat inflamasi lokal
seperti didasari oleh diaktifkannya T-limfosit dengan dominasi suatu dari CD8 + bagian [19]
Sel-sel inflamasi dengan melepaskan sitokin dan faktor pertumbuhan yang dapat
menargetkan sel endotel dapat memberikan kontribusi untuk pengembangan struktural dan
kelainan fungsional dari dinding pembuluh darah [20]
Ada semakin banyak bukti peradangan sistemik pada PPOK seperti yang ditunjukkan
oleh peningkatan tingkat sistemik protein inflamasi seperti protein C-reaktif (CRP) tumor
necrosis factor α (TNF - α) dan interleukin 6 (IL-6) pada pasien ini [21] Baru-baru ini
menunjukkan bahwa pasien PPOK dengan hipertensi pulmonal memiliki serum CRP dan
TNF- α yang lebih tinggi tingkat dibandingkan dengan mereka yang normal tingkat
mPAPnya hal tersebut menunjukkan peran yang mungkin dari peradangan sistemik dalam
patogenesis hipertensi pulmoner [22] Dalam sel endotel manusia CRP mengurangi ekspresi
endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

RPP Pada pasien ini studi post-mortem telah mengungkapkan deposisi otot membujur dan
fibrosis dari intima dari otot arteri pulmoner dengan perkembangan lapisan medial melingkar
otot polos yang dibatasi oleh lamina elastis baru di arteriol
[1213] (Gambar 1)
Gambar 1 Remodeling vaskular pulmoner pada PPOK Catat penebalan intima prominen dan
penyempitan lumen (diambil dari ref [10] dengan ijin)
Remodeling vaskular pulmoner pada penyakit pernapasan biasanya berkembang
dengan adanya hipoksia kronis (PaO2 lt 55-60 mmHg) Dalam studi in vitro telah
menunjukkan bahwa hipoksia menginduksi perubahan dalam produksi dan pelepasan
substansi endotheliumderived vasoaktif proliferasi sel tersebut pada dinding pembuluh darah
serta sintesis peningkatan matriks protein ekstra-seluler Hipoksia yang diinduksi
muskuluarisasi dari prekapiler arteriol yang awalnya dikaitkan dengan VHP persisten
alternatifnya hal tersebut bisa diturunkan dengan transdiferensiasi tersebut sel endotel
menjadi otot polos sebagai respon adaptif dari stres arteri pra-kapiler [14]
Menariknya kelainan pembuluh darah pulmoner terutama yang terdiri dari penebalan
intima dari otot arteri pulmoner oleh proliferasi sel-sel otot polos dan deposisi dari kedua
serat elastis dan kolagen juga telah didokumentasikan pada pasien non-hipoksemia ringan
dan pada perokok dengan fungsi paru-paru normal [15] Pengamatan ini menunjukkan itu
bersama-sama dengan hipoksemia kronis faktor lain bisa memiliki peran dalam patogenesis
HP dan disfungsi endotel dalam fase awal dari PPOK
Penelitian terbaru menunjukkan bahwa kerugian fungsi endotel mungkin timbul dari
perubahan dalam ekspresi dan pelepasan mediator vasoaktif yang penting seperti nitrat
oksida (NO) dan endotelin (ET-1) NO merupakan vasodilator kuat dengan sifat
antiproliferatif yang disintesis oleh NO sintetase (NOS) yang diungkapkan secara konstitutif
pada sel endotel Ekspresi NOS berkurang di arteri pulmonalis Pasien PPOK dengan HP dan
mungkin adalah penyebab dari peningkatan regulasi tersebut dan ekspresi gen yang
mengkodifikasi pada beberapa faktor pertumbuhan seperti faktor pertumbuhan endotel
vaskular [16] Mekanisme ini dapat menyebabkan proliferasi sel dan remodeling pembuluh
arteri pulmonalis ET-1 adalah vasokonstriktor kuat dilepaskan oleh sel endotel yang juga
memberikan suatu efek mitogenik pada otot polos arteri sel ekspresi ET-1 meningkat pada
arteri pulmonalis dari pasien dengan HP hal tersebut menunjukkan peran yang mungkin dari
mediator ini untuk berkontribusi dalam remodeling vaskuler [17]
Adanya perubahan struktural dalam arteri pulmonalis pada perokok dengan fungsi
paru-paru normal dibandingkan dengan non-perokok menunjukkan efek langsung dari
produk rokok-rokok pada struktur pembuluh darab di sisi lain juga diketahui bahwa rokok-
rokok menginduksi disfungsi endotel pada arteri koroner dan sistemik Eksposisi aktif dan
pasif pada asap tembakau mungkin bertindak dengan mengurangi aktivitas dan ekspresi NOS
pada arteri pulmonalis dengan gangguan yang dihasilkan dari fungsi endotel dan remodeling
pembuluh pre-kapiler [18] Merokok juga bisa menyebabkan perubahan inflamasi di arteri
pulmonalis Baru-baru ini peningkatan jumlah infiltrasi sel-sel inflamasi tersebut pada
adventisia arteri pulmonalis pasien dengan PPOK telah dijelaskan infiltrat inflamasi lokal
seperti didasari oleh diaktifkannya T-limfosit dengan dominasi suatu dari CD8 + bagian [19]
Sel-sel inflamasi dengan melepaskan sitokin dan faktor pertumbuhan yang dapat
menargetkan sel endotel dapat memberikan kontribusi untuk pengembangan struktural dan
kelainan fungsional dari dinding pembuluh darah [20]
Ada semakin banyak bukti peradangan sistemik pada PPOK seperti yang ditunjukkan
oleh peningkatan tingkat sistemik protein inflamasi seperti protein C-reaktif (CRP) tumor
necrosis factor α (TNF - α) dan interleukin 6 (IL-6) pada pasien ini [21] Baru-baru ini
menunjukkan bahwa pasien PPOK dengan hipertensi pulmonal memiliki serum CRP dan
TNF- α yang lebih tinggi tingkat dibandingkan dengan mereka yang normal tingkat
mPAPnya hal tersebut menunjukkan peran yang mungkin dari peradangan sistemik dalam
patogenesis hipertensi pulmoner [22] Dalam sel endotel manusia CRP mengurangi ekspresi
endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

Penelitian terbaru menunjukkan bahwa kerugian fungsi endotel mungkin timbul dari
perubahan dalam ekspresi dan pelepasan mediator vasoaktif yang penting seperti nitrat
oksida (NO) dan endotelin (ET-1) NO merupakan vasodilator kuat dengan sifat
antiproliferatif yang disintesis oleh NO sintetase (NOS) yang diungkapkan secara konstitutif
pada sel endotel Ekspresi NOS berkurang di arteri pulmonalis Pasien PPOK dengan HP dan
mungkin adalah penyebab dari peningkatan regulasi tersebut dan ekspresi gen yang
mengkodifikasi pada beberapa faktor pertumbuhan seperti faktor pertumbuhan endotel
vaskular [16] Mekanisme ini dapat menyebabkan proliferasi sel dan remodeling pembuluh
arteri pulmonalis ET-1 adalah vasokonstriktor kuat dilepaskan oleh sel endotel yang juga
memberikan suatu efek mitogenik pada otot polos arteri sel ekspresi ET-1 meningkat pada
arteri pulmonalis dari pasien dengan HP hal tersebut menunjukkan peran yang mungkin dari
mediator ini untuk berkontribusi dalam remodeling vaskuler [17]
Adanya perubahan struktural dalam arteri pulmonalis pada perokok dengan fungsi
paru-paru normal dibandingkan dengan non-perokok menunjukkan efek langsung dari
produk rokok-rokok pada struktur pembuluh darab di sisi lain juga diketahui bahwa rokok-
rokok menginduksi disfungsi endotel pada arteri koroner dan sistemik Eksposisi aktif dan
pasif pada asap tembakau mungkin bertindak dengan mengurangi aktivitas dan ekspresi NOS
pada arteri pulmonalis dengan gangguan yang dihasilkan dari fungsi endotel dan remodeling
pembuluh pre-kapiler [18] Merokok juga bisa menyebabkan perubahan inflamasi di arteri
pulmonalis Baru-baru ini peningkatan jumlah infiltrasi sel-sel inflamasi tersebut pada
adventisia arteri pulmonalis pasien dengan PPOK telah dijelaskan infiltrat inflamasi lokal
seperti didasari oleh diaktifkannya T-limfosit dengan dominasi suatu dari CD8 + bagian [19]
Sel-sel inflamasi dengan melepaskan sitokin dan faktor pertumbuhan yang dapat
menargetkan sel endotel dapat memberikan kontribusi untuk pengembangan struktural dan
kelainan fungsional dari dinding pembuluh darah [20]
Ada semakin banyak bukti peradangan sistemik pada PPOK seperti yang ditunjukkan
oleh peningkatan tingkat sistemik protein inflamasi seperti protein C-reaktif (CRP) tumor
necrosis factor α (TNF - α) dan interleukin 6 (IL-6) pada pasien ini [21] Baru-baru ini
menunjukkan bahwa pasien PPOK dengan hipertensi pulmonal memiliki serum CRP dan
TNF- α yang lebih tinggi tingkat dibandingkan dengan mereka yang normal tingkat
mPAPnya hal tersebut menunjukkan peran yang mungkin dari peradangan sistemik dalam
patogenesis hipertensi pulmoner [22] Dalam sel endotel manusia CRP mengurangi ekspresi
endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

endotel NO sintetase [23] dan meningkatkan rilis ET-1 [24] menunjukkan CRP yang dapat
menyebabkan disfungsi endotel dan berpotensi untuk remodeling vaskular pada sirkulasi
pulmoner TNF- α adalah cytochine pro-inflamasi dengan efek modulator ampuh pada
sirkulasi paru-paru Pada penelitian hewan TNF- α menurunkan produksi prostasiklin pada
sel halus arteri pulmonalis [25] dan meningkatkan plateletactivating factor (PAF) yang
diinduksi vasokonstriksi pulmoner [26] apalagi emfisema dan hipertensi pulmonal berat
berkembang pada tikus transgenik lebih mengekspresikan TNF- α [27]
Pada pasien emfisema hilangnya kapiler pulmoner memainkan peran penting dalam
patogenesis HP Berbeda dengan bentuk HP yang ditandai dengan peningkatan angiogenesis
emfisema luar biasa pada kekurangan relatif pembuluh darah dengan penurunan yang
signifikan dari ekstensi vaskular pulmoner Meskipun mekanisme yang tepat hilangnya
pembuluh paru pada emfisema yang masih belum diketahui telah diusulkan bahwa mungkin
karena efek stres oksidatif dan protease yang menjadi tidak seimbang ketika faktor
pertumbuhan endotel vaskular (FPEV) transduksi sinyal terganggu [28] FPEV tampaknya
menjadi faktor survival wajib bagi mikro-endotel sel vaskular paru mungkin melindungi hal
tersebut terhadap stres oksidatif [29] Pada paru-paru pasien dengan emfisema itu baru-baru
dijelaskan bahwa apoptosis septae alveolar dan sel endotel dikaitkan dengan penurunan
ekspresi FPEV [30] Selain itu dalam model tikus blokade reseptor FPEV menyebabkan
apoptosis sel septum yang mengakibatkan hilangnya kapiler paru-paru dan emfisema dan
menyebabkan beberapa derajat HP [31] Sangat baru-baru ini penelitian menunjukkan bahwa
kerusakan pembuluh darah pulmonalis yang diamati dalam PPOK juga bisa disebabkan oleh
berkurangnya produksi dari sumsum tulang sel progenitor endotel sirkulasi (SPES) di
samping itu berkurangnya jumlah SPES tampaknya berkorelasi dengan tingkat keparahan
penyakit itu [3233]
23 Hiperinflasi Paru
Hiperinflasi paru adalah mekanisme patofisiologis yang merupakan penyebab penting
dari dispneu dan intoleransi olahraga pada PPOK Pasien dengan PPOK dan khususnya yang
emphysematous menunjukkan peningkatan volume paru-paru statis (misalnya volume
residu kapasitas fungsional residu jumlah volume paru) sebagai akibat dari obstruksi kedua
bronkus yang mengurangi aliran napas ekspirasi yang menghambat pengosongan lengkap
paru dan hilangnya parenkim pulmonal yang mengurangi elastisitas dari paru hiperinflasi
paru lebih jelas selama latihan fisik (hiperinflasi dinamik) karena peningkatan tingkat
pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

pernapasan dan selanjutnya pengurangan waktu ekspirasi diperlukan untuk deflasi pulmoner
Hiperinflasi dinamis mungkin memainkan peran penting dalam patogenesis olahraga terkait
HP dan gagal jantung kanan Pada volume pulmoner tinggi dinding alveolar tersebut
meregang dan menyebabkan kompresi kapiler alveolar tersebut dengan peningkatan RPP dan
arteri tekanan pulmonal [34] Selain itu hiperinflasi meningkatkan tekanan intratoraks
juxtacardiac yang menyebabkan kenaikan dari tekanan atrium kanan pengurangan
konsekuen gradien balik vena tersebut dapat membahayakan pengisian ventrikel kanan dan
menyebabkan penurunan kinerja jantung [35]
Semua mekanisme yang disebutkan di atas mungkin bervariasi memberikan
kontribusi pada patogenesis HP pada PPOK dan aksi terintegrasi tersebut mmungkin
menjelaskan mengapa bahkan jika ringan saat istirahat HP mungkin meningkat selama
latihan fisik Pada pasien PPOK berat dengan HP istirahat kenaikan mPAP dari 27 mmHg
sampai 55 mmHg yang dilaporkan selama 30-40 W latihan keadaan stabil dari 7-10 menit
durasi [36] Pada orang normal peningkatan fisiologis curah jantung selama latihan diimbangi
oleh penurunan proporsional RPP sehingga mPAP tetap dalam kisaran normal pengurangan
RPP adalah karena vasodilatasi pembuluh paru dan perekrutan kapiler biasanya tidak perfusi
Pada pasien PPOK kompensasi mekanisme ini kurang efisien karena remodeling vaskular
pulmoner dan hilangnya pembuluh pulmonalis Latihan dapat meningkatkan VHP karena
mengurangi campuran vena PO2 meningkatkan tonus sistem saraf simpatik dan mengurangi
HP arteri Latihan yang diinduksi hiperinflasi paru-paru juga dapat bertindak mempengaruhi
resistensi kedua pembuluh darah pulmonal dan mengubah kinerja jantung kanan meskipun
HP dapat berkembang pada intensitas latihan moderat kemungkinan bahwa episode berulang
dari HP yang diinduksi latihan bisa benar-benar memberikan kontribusi pada pengembangan
hipertrofi ventrikel dan kemudian kegagalan ventrikel kanan tetap spekulatif
3 RENAL DAN KELAINAN ABNORMAL PADA KOR PULMONAL KRONIK
Pada tahun 1960 Campbell dan Short sudah menunjukkan bahwa pasien dengan
PPOK edema perifer kor pulmonal hampir selalu terkait dengan retensi karbon dioksida
(CO2) [37] Telah diusulkan CO2 itu baik langsung atau melalui jalur humoral
mengaktifkan retensi natrium dalam ginjal mungkin bekerja pada natrium - hidrogen yang
(Na + H +)-antiporter dalam membran luminal tubulus proksimal yang terlibat dalam
buffering pernapasan asidosis dengan mengorbankan keuntungan natrium dalam tubuh [4]
Hal ini juga ditetapkan bahwa dengan adanya hiperkapnia perfusi ginjal semakin jatuh dan
hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

hubungan terbalik yang signifikan antara arteri karbon dioksida ketegangan (PaCO2) dan
aliran plasma ginjal telah berulang kali dijelaskan [4]
(Gambar 2)
Gambar (2) Hubungan antara aliran plasma renal (RPF) dan Tekanan arterial karbon
dioksida (PaCO2) pada pasien PPOK (diambil dari ref [4] dengan ijin)
Selama dekade terakhir fungsi ginjal pada pasien PPOK telah ditandai dengan baik
dan telah muncul bahwa aliran plasma ginjal efektif (ERPF) berkurang dalam lanjutan fase
PPOK khususnya selama eksaserbasi akut dan atau ketika edema perifer hadir [38]
Meskipun penurunan aliran darah ginjal laju filtrasi glomerulus (GFR) biasanya
dipertahankan sampai fase akhir dari penyakit sesuai fraksi filtrasi (FF) terus meningkat pada
PPOK yang memburuk [38] Kenaikan FF reabsorpsi proksimal natrium dan air karena
tekanan hidrostatik peritubular lebih rendah dan tekanan onkotik lebih tinggi bila sebagian
besar dari cairan glomerular disaring [3] Menariknya curah jantung biasanya normal
bahkan dalam PPOK berat termasuk bahwa penurunan ERPF sekunder pada penurunan
kinerja jantung tetapi menunjuk ke arah adanya vasokonstriksi wilayah ginjal
Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

Meskipun mekanisme belum sepenuhnya dijelaskan korelasi erat antara PaCO2
ERPF dan ekskresi natrium menunjukkan bahwa hiperkapnia mungkin menjadi faktor yang
bertanggung jawab untuk vasokonstriksi ginjal hal ini didukung oleh pengamatan bahwa
ketika hiperkapnia mempersulit PPOK respon vasodilator dosis rendah dopamin dan jangka
pendek oksigen hilang [39] Hiperkapnia dapat menyebabkan vasokonstriksi langsung
maupun tidak langsung dengan merangsang tonus simpatik seperti yang disarankan oleh
peningkatan tingkat sirkulasi dari katekolamin pada pasien edema dengan PPOK berat [38]
Tonus simpatik lebih meningkatkan reabsorpsi Na+ tubular dengan mengurangi ERPF dan
atau mendistribusikan kembali aliran darah ginjal
Hipoksemia tanpa hiperkapnia bersamaan tampaknya tidak mampu mendorong
perubahan signifikan dalam hemodinamik ginjal dan atau Na+ dan homeostasis air
meskipun pemberian oksigen tampaknya mengerahkan efek vasodilator ginjal pada pasien
PPOK normokapnik hipoksemia [39] dalam pasien hiperkapnia nonoedematous yang
membutuhkan tambahan oksigen jangka panjang memburuknya hipoksemia menyebabkan
signifikan jatuhnya output natrium urin melalui penurunan laju filtrasi glomerulus [40]
Selain itu koreksi hipoksemia dengan terapi oksigen jangka panjang pada pasien hiperkapnia
dengan hasil kegagalan pernafasan kronis yang ditingkatkan natriuresis menunjukkan
hipoksemia itu di hadapan hiperkapnia memberikan kontribusi untuk retensi natrium dalam
PPOK [41]
Seperti disebutkan sebelumnya tingkat katekolamin plasma meningkat sebagai
respon terhadap retensi CO2 sejak fase awal PPOK aktivasi simpatik merangsang
reninangiotensin- aldosteron dan meningkatkan penyerapan tubular bikarbonat natrium dan
air menyebabkan vasokonstriksi ginjal dan retensi Na+ Disebutkan mekanisme di atas bisa
menjelaskan episode air dan retensi Na+ selama eksaserbasi PPOK yaitu memburuknya
pertukaran gas paru yang umumnya menyebabkan kegagalan pernapasan dan hiperkapnia
Bukti eksperimental menunjukkan bahwa dalam PPOK lanjutan kemampuan untuk
mengeluarkan Na + dan air lebih lanjut diperburuk oleh aktivasi sistem aktivitas renin plasma
dan aldosteron plasma ( PRA - PA ) Dalam banyak pasien edema tingkat sirkulasi tinggi
renin angiotensin II dan aldosteron diamati dengan nilai 2-3 kali lebih tinggi dibandingkan
subyek normal tetapi biasanya lebih rendah daripada yang diamati pada gagal jantung
kongestif [ 42 ] Dalam PPOK PRA - PA dirangsang hanya bila GFR mulai jatuh sebagai
akibat dari penurunan signifikan ERPF [ 43 ] hal tersebut menunjukkan bahwa aktivasi
sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

sumbu PRA - PA tergantung pada aktivasi apparatus juxtaglomerular yang disebabkan baik
oleh penurunan aferen aliran arteri ginjal dan dengan mengurangi Na + yang disaring
bebannya Meskipun aktivitas PRA ndash PA berkorelasi dengan kemampuan ginjal untuk
mengeluarkan natrium hal itu tidak mungkin untuk menetapkan dengan pasti berapa banyak
korelasi ini tergantung pada berkontribusi nyata aktivasi PRA - PA pada retensi Na + daripada
hanya pengurangan ERPF Di sisi lain hasil mengenai kapasitas angiotensin converting
enzyme inhibitor untuk meningkatkan ekskresi natrium PPOK edema pasien itu kontroversial
[ 44 ]
Hiponatremia signifikan hadir dalam cukup jumlah pasien PPOK edema mereka
umumnya memiliki peningkatan tingkat arginine vasopressin (AVP) yang tidak tepat
tingginya untuk nilai osmolalitas plasma menunjukkan bahwa mekanisme non-osmotik dapat
mengatur produksi AVP di PPOK [45] Seperti dalam kegagalan pernapasan kronis Pasien
PPOK edema telah meningkatkan tingkat ANP [46] yang dilepaskan dari dinding atrium
yang distended dan biasanya bertindak sebagai mekanisme perlindungan edema karena
adanya vasodilator diuretik dan faktor natriuretik Sebaliknya pada subjek normal pada
pasien PPOK peningkatan ANP tampaknya tidak mengerahkan efek penekan dalam sistem
PRA-RA pada pasien ini surpresi PRA-PA ada yang mempromosikan Na + dan ekskresi air
Ini mungkin berarti bahwa pada pasien PPOK edema efek dari pengurangan ERPF pada rilis
PRA-RA adalah lebih kuat daripada efek AVP pada supresi PRA-RA
Adanya peningkatan tekanan ekspirasi intrathoraks seperti yang diamati pada pasien
PPOK selama latihan atau selama eksaserbasi penyakit bisa berkontribusi pada kelainan
ginjal dan hormonal diamati dalam perjalanan penyakit Telah dilaporkan bahwa tekanan
positif pernapasan menurunkan aliran urin yang dihasilkan terutama dari penurunan di
pembersihan air bebas dengan penurunan GFR sebagai faktor [47] Retensi air juga bisa
disebabkan oleh kelebihan produksi ADH yang dihasilkan oleh efek peningkatan tekanan
intratoraks pada reseptor peregangan yang ada pada dinding atrium dan pembuluh yang lebih
besar dari thorax [48] Penelitian lebih lanjut bagaimanapun juga diperlukan untuk
memperjelas kemungkinan interaksi yang kompleks antara mekanika paru (dan pertukaran
gas) dan fungsi ginjal pada PPOK
Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

Fig (3) Patogenesis hipertesi pulmoner and ldquoKor Pulmonalrdquo kronis in PPOK Garis tebal menunjukan mekanisme yang ada
garis putus - putus menunjukkan hal ndash hal yang belum dibuktikan PRA=plasma renin activity PA=plasma aldost
4 PERSPEKTIF MASA DEPAN
Dalam dekade terakhir pertanyaan jika Kor pulmonal kronis benar-benar ada telah
diangkat dan terjadinya kegagalan jantung kanan dalam PPOK dipertanyakan Bukti anatomi
kanan hipertrofi ventrikel dapat ditemukan di otopsi di atas 40 dari pasien PPOK dengan
beberapa perbedaan antara seri yang berbeda yang disebabkan oleh kurangnya kriteria
morfologi terpadu yang digunakan untuk mendefinisikan hipertrofi ventrikel umumnya
pada pasien PPOK stabil stroke volume ventrikel yang tepat dipertahankan dengan
menggunakan penilaian enddiastolic hubungan tekanan yang memungkinkan evaluasi
lebih akurat kontraktilitas ventrikel kanan dibandingkan untuk perhitungan fraksi ejeksi
ventrikel kanan yang telah ditunjukkan bahwa kontraktilitas dan output jantung dari
ventrikel kanan biasanya ada dalam pasien stabil [ 49 ] Beberapa pertimbangan tambahan
bagaimanapun juga itu dibutuhkan Peningkatan yang signifikan dari tekanan arteri paru
telah dijelaskan selama latihan pada PPOK dan mungkin itu sementara dapat mengurangi
kinerja ventrikel kanan dengan tidak adanya disfungsi jantung istirahat yang signifikan
Kurangnya evaluasi yang ekstensif fungsi jantung kanan selama latihan tidak memungkinkan
pembentukan dampak nyata dari HP pada kinerja jantung pada pasien PPOK Selain itu efek
lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

lain mekanisme intermittent seperti desaturasi oksigen arteri selama latihan dan atau
tidur pada fungsi jantung kanan perlu dipelajari
Edema perifer sering diamati pada hiperkapnia Pasien PPOK terutama selama fase
eksaserbasi Peran tekanan pulmoner yang berlebihan dalam pengembangan gagal jantung
kanan telah diperdebatkan karena kurangnya korelasi antara nilai mPAP dan kehadiran
edema perifer dan atau tanda-tanda hemodinamik gagal jantung kanan Pengamatan ini
bersama-sama dengan akuisisi baru tentang perubahan ginjal dan hormonal dalam pasien
PPOK hal tersebut menyebabkan asumsi bahwa pembentukan edema PPOK lanjutan
sebagian besar disebabkan adanya retensi natrium dan air yang disebabkan oleh asidosis
hiperkapnia dan diperburuk oleh hipoksemia berat Ginjal tidak berasal jantung dari edema
perifer menjelaskan mengapa pasien emphysematous ( Pink Puffers ) yang dapat
memiliki nilai yang lebih tinggi sebagai mPAP dibandingkan dengan pasien bronkitis kronis (
blue bloaters ) biasanya tidak mengalami retensi air dan natrium bahkan selama fase
lanjut dari penyakit Pengaruh intermiten hipoksia intermiten dan juga hiperoxia pada fungsi
ginjal dan pelepasan hormon belum diselidiki pada PPOK Ada bukti bahwa pada pasien
dengan obstructive sleep apnea hipoksia intermiten dapat menyebabkan tidak hanya
pelepasan katekolamin [ 50 ] tetapi juga rilis sitokin pro - inflamasi [ 51 ] Studi masa
depan diperlukan untuk memverifikasi apakah jalur ini juga berlaku pada pasien dengan
PPOK dan apa yang disebut kor pulmonal (Gambar 3 )
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013
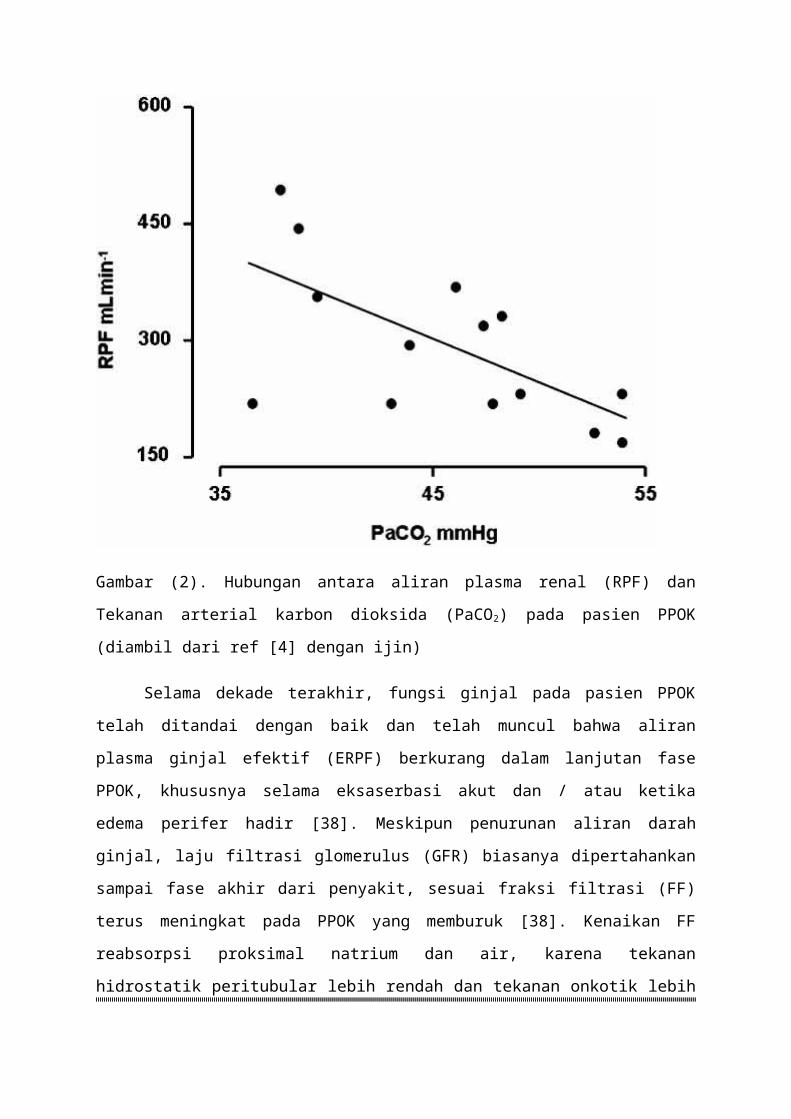
Tugas Ujian Prof Dr Suradi dr MARS
PATOGENESIS KRONIS KOR PULMONAL PADA PPOK
(Pathogenesis of Chronic ldquoCor pulmonalerdquo in COPD)
Paolo Palange dan Patrizia Paoletti
OLEH
Fahmi Wahyu R G99112068
PENGUJI
Prof Dr Suradi dr MARS
KEPANITERAAN KLINIK SMF ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN UNS RSUD DR MOEWARDI
SURAKARTA
2013

2013




















